
Can Your Risk Management Prevent a Recall?
Recalls are costly and damage trust in your brand. Effective risk management can help.

A thermodilution catheter used to quickly measure blood pressure, flow and oxygen levels during complex life-saving surgeries is recalled due to incorrect assembly and reversal of catheter tubes, which may cause inaccurate readings, leading to potentially unintended treatment.
A safety guard used with a ventilator to provide breathing support to infants and children is recalled due to a manufacturing defect, which may cause the device to disconnect from the patient’s breathing circuit.
A defibrillator used to deliver a life-saving electrical shock in the event of cardiac arrest is recalled due to a design issue, which causes unexpected power failure during use, potentially leading to a risk of serious injury or death due to delay in therapy.
These are examples of recent Class I recalls, the most serious type of recall classification by the FDA, where continued use of, or exposure to, these devices is reasonably expected to cause serious injury or death. In the three examples cited above, there is clearly a very high risk of serious injury or death due to product defects. Class II and Class III are the other two types of recall classifications corresponding to lower risk levels.
Recalls are quite common in MedTech, but its only now that public attention on medical device safety is rising. In late 2018, Associated Press published a story that reported more than 1.7 million injuries and nearly 83,000 deaths suspected of being linked to medical devices reported to the FDA over a 10-year period. Soon after that Netflix released The Bleeding Edge, a documentary which captured public attention through personal stories of patients harmed by routinely implanted medical devices. Media coverage has increased significantly since then, and there is a news story on TV or in the print media almost on a weekly basis. Although the shock value of these stories is not as high as the Boeing 737 Max crashes, which led to an indefinite grounding of these aircrafts worldwide, the truth is that millions of patients are exposed to significant risks on a daily basis by unsafe medical devices.
There is a lot of publicly available FDA data that can be analyzed to better understand patterns linked to system level issues with medical devices. Recently, we analyzed this data and summarized our findings in an infographic Medical Device Recalls (2015-Apr 2019). Here are a few key highlights:
Over 90% of the nearly 4650 recall events reported since 2015 are classified as Class II. However, Class I recalls reflect a disproportionate increase from 3-4% a year to nearly 6% of recall events reported in 2019 through the month of April.
Top 5 medical specialties reflecting nearly 60% of total recall events since 2015 are: Radiology (16%), Orthopedic (13%), Cardiovascular (11%), General Plastic Surgery (10%) and General Hospital (9%).
Cardiovascular medical specialty is the largest contributor to Class I recalls representing 27% of the 174 recall events since 2015. Next two medical specialties with high rates of Class I recalls are Anesthesiology (21%) and General Hospital (7%).
Radiology is the largest contributor to Class II recalls representing 17% of the 4265 recall events since 2015. Next two medical specialties with high rates of Class II recalls are Orthopedic (14%) and General Plastic Surgery (11%).
Cardiovascular and Orthopedic specialties are the two of the largest medical device segments representing a combined 23% of the overall industry sales. It is likely that we all know of someone who with a hip or knee replacement, or a stent implanted to keep an artery open. There is no doubt that medical devices help save lives and have made a significant contribution to public health and quality of life. Yet we cannot underestimate the public impact of so many recalls in the industry.
The data shows us key areas where we must focus our efforts. As shown in Exhibit 1, nearly 25% of the recall events are due to either a Device Design or Software Design issue. Process Control issues, including release of Non-conforming materials and components are also common. Even if you have not had a recall, or your products are not in the top medical specialties cited above, there is a good chance your Quality Management System (QMS) is also vulnerable to deficiencies in these areas.
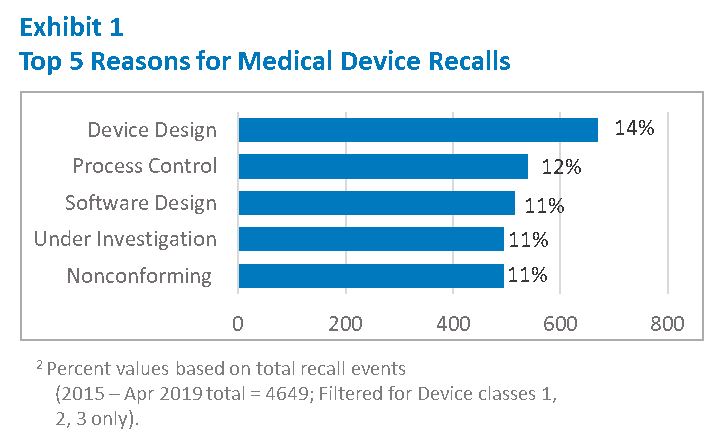
An effective QMS is required by regulations to ensure continued safety and effectiveness of medical devices. These requirements have been in place for a very long time and manufacturers have implemented multiple safeguards in the system, including the required internal audits to identify and correct systemic quality issues. Yet the frequency of recalls and top reasons highlighted in Exhibit 1 suggest continued systemic deficiencies in the QMS.
When it comes to Design, there is a full set of requirements in FDA regulations, appropriately labeled Design Control. It was nearly 30 years ago that the Safe Medical Device Act authorized FDA to add Design Control to the current Good Manufacturing Practice (cGMP) requirements for medical devices. Later, these requirements were further formalized in the Quality System Regulation (QSR), which replaced the 1978 GMP requirements. It can be argued that the industry has had a lot of time to improve Design Controls.
Why does MedTech continue to struggle with Design? What are the barriers to good, reliable design of medical devices?
Our recent article on LinkedIn, MedTech has a Design Problem, resulted in a very robust discussion on this topic, and a few insightful comments led to several top-level themes:
Risk management during design and development
Limited or lack of, understanding of, Design Control principles
Making assumptions that design changes do not impact performance against requirements
Cultural barriers where Quality is not perceived to be owned by all
Excessive focus on documentation compliance
A common thread running through these perceived barriers is risk management and how it is practiced in the industry. ISO 14971 is the recognized international standard that defines the requirements of risk management for medical devices, and is widely applied in the industry. The challenge lies in effectively integrating these requirements across the QMS in an efficient way to manage risks throughout the product lifecycle. It is easier said than done, and most organizations find it challenging to link various sub-systems of their QMS in a way to maintain clear oversight and control of risk. There is no shortage of forms, checklists and templates being used in the industry for “managing” risk; however current practices generally focus on compliance and have failed to provide effective risk management.
What is “effective” risk management?
Risk management is effective when it is integrated across the QMS in a consistent way. It starts with a clear policy, supported by procedures linking different sub-systems through consistent, non-contradictory requirements. When evaluating the effectiveness of your risk management system, it is useful to consider the following questions:
Do we have a common understanding of risk? As an example, risk of product failure is not the same as risk of harm to patients and users.
Do we have a good understanding of harms that can be caused by the use of, or exposure to, our product? Do we clearly understand the different ways, or sequence of events, which may create a hazardous situation for our patients and users?
Do we have a standardized way to evaluate risk of these harms and have we established patient-centric criteria for risk acceptability to be used across the QMS?
Are we using the right tools for analysis and evaluation of risks?
Do we have clear criteria for evaluation of required controls and their effectiveness? Do these controls continue to be effective when changes are made?
Do we have the ability to accurately detect signals with a potential to affect current risks or introduce new risks? How do we separate noise from a true signal?
Do we have the ability to take preventive measures when these signals are detected?
Is our Quality System sufficiently responsive to changes, both internal and external?
Risk management needs to be treated as a business process, not an exercise in Compliance. Just the way business and financial risks are carefully managed, product quality and safety risks also need to be diligently managed by Executive Leadership. Decision to recall a product due to safety concerns is not an easy one, and leads to difficult, often contentious, discussions in the Board Room. It is not uncommon for the top executives to be surprised when confronted with a situation where such decisions need to be made. “How could we have missed this?”, they ask their Safety and Quality leaders, who in turn, rarely have good answers. Generally, the product quality and safety issues are not considered high business risks just because “we haven’t had any FDA issues”.
In fact, recalls and other chronic quality issues in the field are a significant business risk in real financial terms. According to a recent McKinsey report, non-routine external quality failures such as recalls, are estimated to cost as much as 3.8% of industry sales, or about $14.4B. The true cost of a serious recall is more than just a financial impact to the bottom line. Public loss of confidence in the product, and the brand, can have a far greater impact on the viability of the business itself.
It is not enough to ask “do we have a risk management system?”; we need to ask if our risk management system is truly protecting our patients from harm, and our business from financial ruin.
It’s time to have high expectations of your risk management process!
References:
AP New Story – How Global Journalists Investigated Medical Device Safety, November 2018
The Bleeding Edge – a Netflix Documentary, 2019
Boeing 737 Max Crashes – 27 Billion Reasons to Get Serious About Risk Management , March 2019
FDA Recalls Classification, April 2019
McKinsey Report – Capturing the Value of Good Quality in Medical Devices, February 2017
MedTech has a Design Problem, May 2019
Infographic – Medical Device Recalls (2015 – April 2019), May 2019